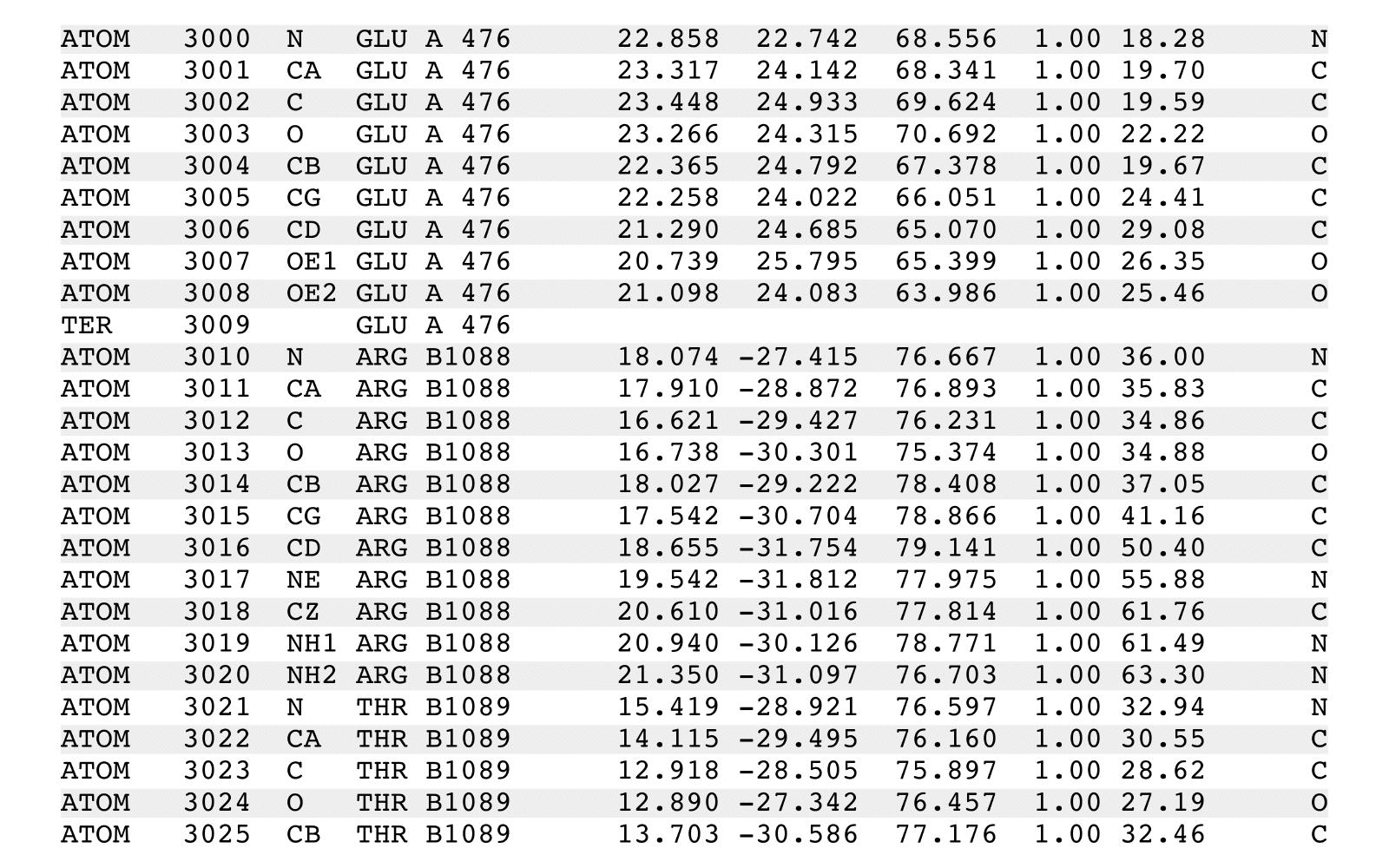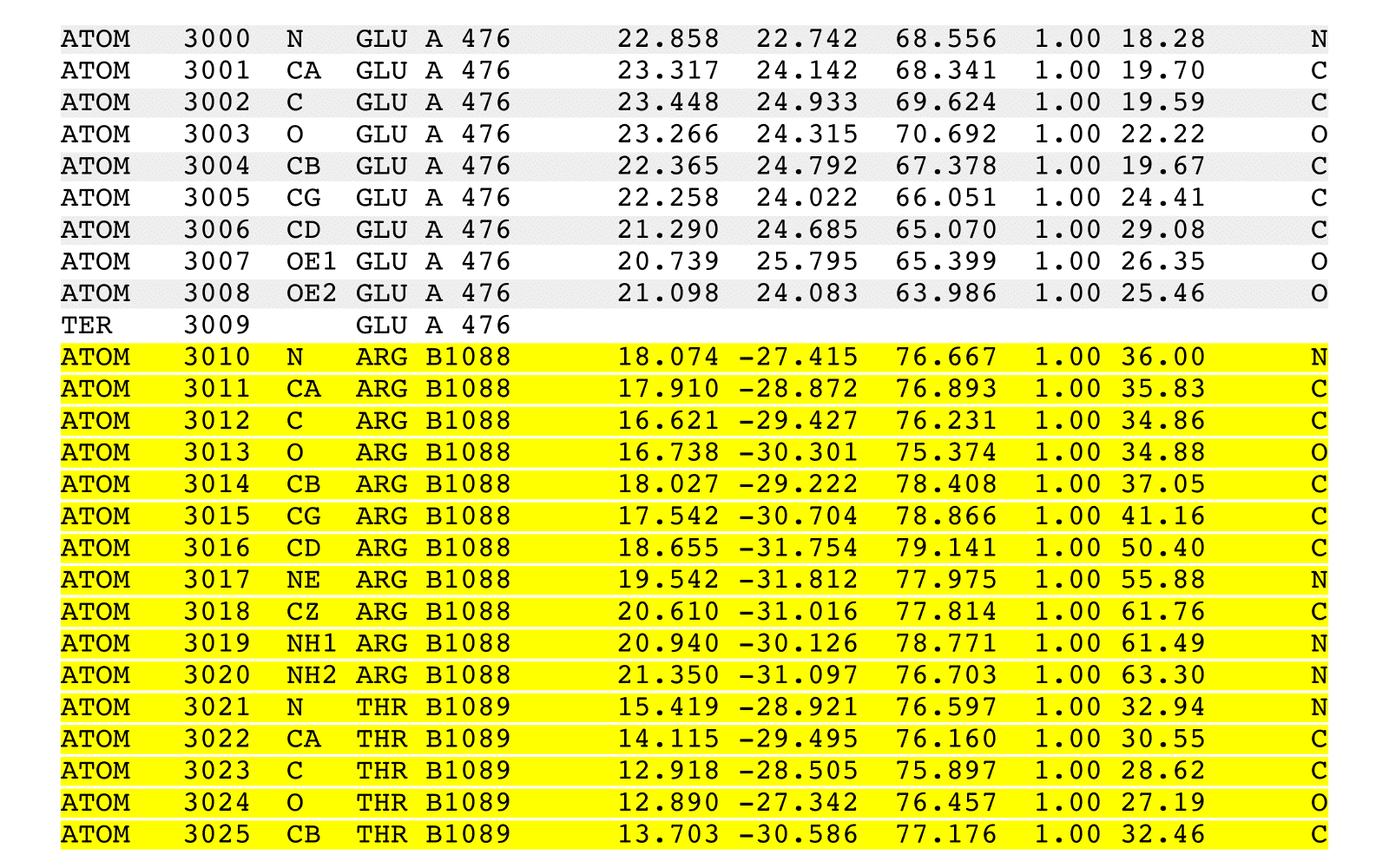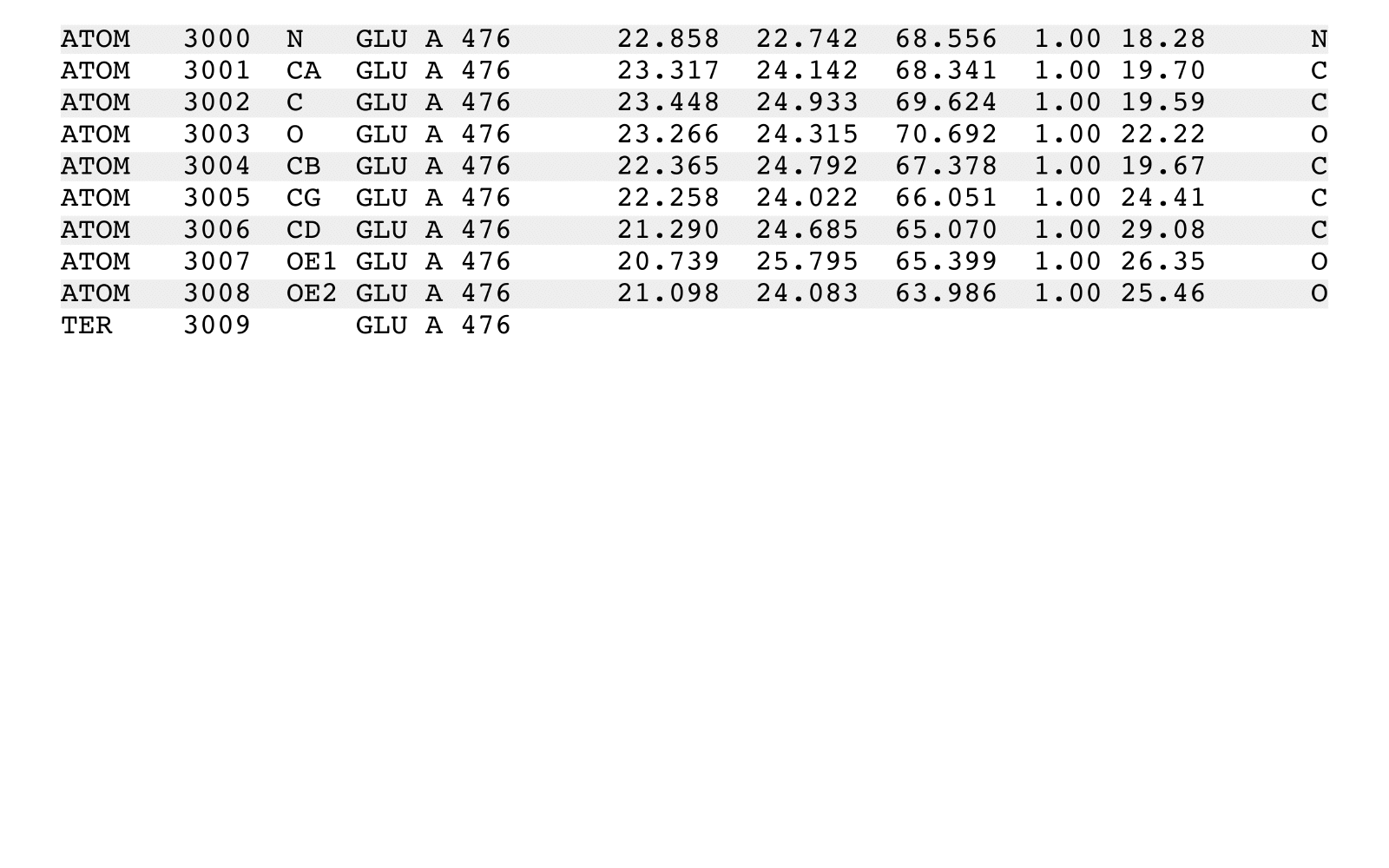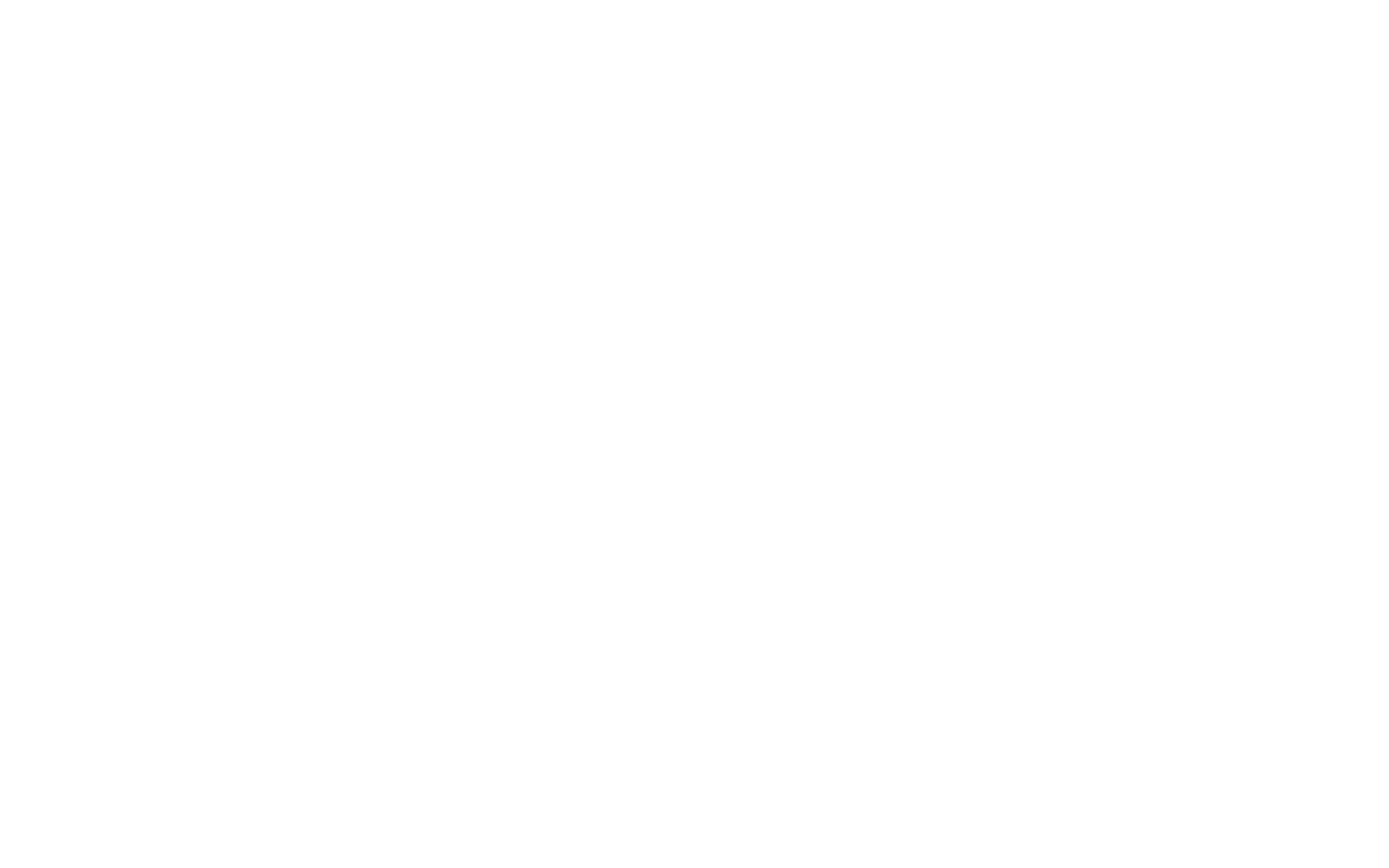
stripPDB
Overview
stripPDB Help Output
stripPDB Tutorial
(WEB PAGE PDF)
stripPDB Overview
stripPDB is a PDB filter to strip specified atoms.
stripPDB Help Output (“stripPDB -h” output)
NAME
stripPDB (version 1.0.2) -- a PDB filter to strip specified atoms
SYNOPSIS
stripPDB [options]
CHARACTER OPTION____KEYWORD OPTION________DESCRIPTION________________________DEFAULT____
-i <filename> ... --input=<filename> .... input PDB file ................... stdin
-o <filename> ... --output=<filename> ... output PDB file .................. stdout
-m <model#> ..... --model=<model#> ...... MODEL number of input PDB file ... first model
-r <remove_str> . --remove=<remove_str> . remove atoms w/specific chain IDs. no action
<remove_str> is either: <chain_ids> (simple list of chain IDs w/no unquoted ';' or ',')
or: [[c],[r],[a];]+ (triplet list of chain ID, residue #, atom #)
-t .............. --hetatms ............. remove all HETATMs ............... no action
-w .............. --water ............... remove all HETATM water atoms .... no action
-y .............. --hydrogen ............ remove all HETATM hydrogen atoms . no action
-h .............. --help ................ prints help (Enter 'stripPDB -h' for more help!)
<NO OPTIONS> ............................ shorter option synopsis (Enter 'stripPDB'.)
--license ............. prints license terms for stripPDB.
DESCRIPTION
stripPDB removes specified atoms from an input PDB file and writes a new output PDB file.
stripPDB will process one (possibly implied) MODEL from the input PDB file. The output
PDB file will contain: (i) the header records from the input file, (ii) additional REMARK
header records summarizing stripPDB execution, (iii) all the records from a single specific
MODEL with the exception of any ATOM, HETATM, and TER records that match removal criteria,
(iv) any CONECT records that reference output records, and (v) a final END record.
Records of any other existing MODEL sections, and their associated CONECT records,
will be removed. CONECT records will be updated to reflect removed records.
The input file is expected to be in PDB format. Option '--input=' ('-i') specifies the
input filename. If option '--input=' ('-i') is not present, input will be read from stdin.
By default, the first MODEL (perhaps implicit if no MODEL records exist) from each
input file is processed. Option '--model=' ('-m') may be used to specify the MODEL
number of the input file to process.
Option '--output=' ('-o') specifies the output filename. If option '--output=' ('-o')
is not present, output will be written to stdout.
Option '--hetatms' ('-t)' specifies that all HETATM records are to be removed.
Option '--water' ('-w') specifies that all HETATM records that are also water atoms
(hydrogen or oxygen) are to be removed. Water atoms are defined as HETATM records
that have a Residue Name value of "HOH".
Option '--hydrogen' ('-y') specifies that all HETATM records that are also hydrogen
atoms (regardless of whether they are part of water) are to be removed.
If the option '--remove=' ('-r') is present, then any ATOM, HETATM, and TER record
having content that matches the target specification of the '--remove=' ('-r') option
value will be removed. The '--remove=' ('-r') option value is either a simple list of
chain identifiers ("Simple List Format") or it is a list of triplets that allow chain
identifiers, residue sequence numbers, and atom serial numbers to be used to specify
matching record targets ("Triplet List Format").
Simple List Format
The Simple List Format of the '--remove=' ('-r') option value is a character string of
chain identifiers. This character string can be any size and can contain any non-NULL
character. If this character string starts and ends with either a single quote (''')
or a double quote ('"'), then these quotes will be treated as string delimiters and not
as chain indentifiers. When quotes are intended to be used as chain indentifiers, then
the quote characters must be preceded with a single backslash character ('\'). This
character string may not contain semicolons (';') or commas (',') unless the semicolons
and commas are preceeded with a backslash (e.g., '\;'); semicolons and commas not quoted
by a backslash will cause the '--remove=' ('-r') option value to be interpreted as a
Triplet List Format. Two consecutive backslashes ('\\') are used when the backslash
character ('\') is intended to be used as a chain identifier (i.e., the backslash
character that is the chain identifier gets quoted with a backslash).
Examples of the Simple List Format follow:
Terse option Keyword option Description
-------------- ----------------- --------------------------------------
-r ABC --remove=ABC chain IDs 'A','B','C'
-r 'ABC XYZ' --remove='ABC XYZ' chain IDs 'A','B','C',' ','X','Y','Z'
-r "ABC AAA" --remove="ABC AAA" chain IDs 'A','B','C',' ','A','A','A'
Note that a single quote (') or a double quote (") may be used to delimit an option value.
The '--remove=' ('-r') option values shown above are quoted in order to specify that a
space (' ')be used as a character identifier.
Note that use of the hyphen character ('-') in the Simple List Format will be interpreted as
the value of a chain identifier. See “Triplet List Format” below for range specifications.
Triplet List Format
The Triplet List Format of the '--remove=' ('-r') option value allows records to be specified
for removal by presence of chain identifier, residue sequence number, and atom serial number.
This triplet list is defined as a semicolon-delimited list of triplets, where each triplet
consists of three comma-delimited fields:
chain identifier, residue sequence number, atom serial number
Any of the '--remove=' ('-r') triplet fields may optionally have a range specification
in one of the following formats:
Format Examples Description
----------------------------- -------- --------------------------------------------
<lower_limit> - <upper_limit> A-Z Matches any value in between and including
107-200 lower limit and upper limit.
- <upper_limit> -Z Matches any value lower than and including
-200 upper limit.
<lower_limit> - A- Matches any value higher than and including
107- lower limit.
Any of the '--remove=' ('-r') triplet fields may have a null value (i.e., an empty field).
Null-valued triplet fields will match any record value of that field type. A PDB record
has to match all non-null fields in a triplet in order for record removal.
Comma delimiters need only be supplied as needed to specify a field index. Semicolon
delimiters are required with the exception of the last semicolon, which is optional.
Examples of the triplet list format follow:
Terse option Keyword option Description
-------------------- -------------------------- -------------------------------------------
-r 'A;B;C;' --remove='A;B;C;' chain IDs (any residue # and any atom #)
-r 'A;B;C' --remove='A;B;C' chain IDs (any residue # and any atom #)
-r 'A;B;C; ;X;Y;Z' --remove='A;B;C; ;X;Y;Z' chain IDs (any residue # and any atom #)
-r "A;B;C; ;A;A;A" --remove="A;B;C; ;A;A;A" chain IDs (any residue # and any atom #)
-r "A,,;" --remove="A,,;" chain ID 'A' (any residue # and any atom #)
-r "A,35,;" --remove="A,35,;" residue 35 of chain ID 'A' (with any atom #)
-r "A,35,293;" --remove="A,35,293;" atom 293 of residue 35 of chain ID 'A'
-r ",35,;" --remove=",35,;" residue 35 (any chain ID and any atom #)
-r ",,293;" --remove=",,293;" atom 293 (any chain ID and any residue #)
-r "A,,;,35,;,,293;" --remove="A,,;,35,;,,293;" three triplets with complete delimiters
-r "A;,35;,,293" --remove="A;,35;,,293" three triplets with necessary delimiters
-r ",-78;,110-;" --remove=",-78;,110-;" all residue #s except 79 through 109
Notes for both Simple List Format and Triplet List Format
The '--remove=' ('-r') target specification consists of single characters (representing chain
identifiers) for the Simple List Format and triplets (providing more flexible specifications)
for the Triplet List Format.
Chain identifiers, specified in '--remove=' ('-r') option values, can be any non-NULL character.
If semicolons (';'), commas (','), backslashes ('\'), or option value quotes (''' or '"') are
used as chain identifier values in any '--remove=' ('-r') option value, then these characters
need to be preceded with two consecutive backslashes ('\\').
As the unix shell interprets semicolons (';') as command line separators, command line option
values containing semicolons (';') are best handled by quoting the entire option value with
either single quotes (') or double quotes (").
New "REMARK 250" lines will be added to the output PDB specifying execution details.
By default, no PDB records are altered or removed. Options '--hetatms' ('-t)', '--water' ('-w'),
'--hydrogen' ('-y'), or '--remove=' ('-r') all independently specify PDB records to be removed
(i.e., any record specified to be removed by any of these options will be removed regardless
of the use or value of any other option.) If no removal options are provided, then stripPDB
will make a copy of the entire PDB (including all MODELS).
Any errors and warnings will be written to stderr.
EXAMPLE
The following will remove all HETATM records (and any associated CONECT records)
from 'in.pdb'. The file 'out.pdb' will be created.
With keyword options:
stripPDB --input=in.pdb --output=out.pdb --hetatms
With character options:
stripPDB -i in.pdb -o out.pdb -t
The following will remove all records (and any associated CONECT records)
from 'in.pdb' that contain either 'P' or 'Q' as a chain identfier.
The file 'out.pdb' will be created.
With keyword options:
stripPDB --input=in.pdb --output=out.pdb --remove=PQ
With character options:
stripPDB -i in.pdb -o out.pdb -r PQ
The following will remove all records (and any associated CONECT records) from
'in.pdb' that contain either 'P' or 'Q' or space (' ') as a chain identfier.
The file 'out.pdb' will be created.
With keyword options:
stripPDB --input=in.pdb --output=out.pdb --remove="PQ "
With character options:
stripPDB -i in.pdb -o out.pdb -r "PQ "
With keyword options in a Triplet List Format:
stripPDB --input=in.pdb --output=out.pdb --remove="P;Q; ;"
With character options in a Triplet List Format:
stripPDB -i in.pdb -o out.pdb -r "P;Q; ;"
The following will remove all records (and any associated CONECT records) from
'in.pdb' that contain both the chain identifier 'A' and the residue number 36.
With keyword options:
stripPDB --input=in.pdb --output=out.pdb --remove="A,36"
With character options:
stripPDB -i in.pdb -o out.pdb -r "A,36"
The following will remove all records (and any associated CONECT records) from
'in.pdb' that contain the residue sequence number 35 (and any chain identifier).
With keyword options:
stripPDB --input=in.pdb --output=out.pdb --remove=",35"
With character options:
stripPDB -i in.pdb -o out.pdb -r ",35"
The following will remove all records (and any associated CONECT records) from 'in.pdb'
having the atom serial number 293 (regardless of chain identifier or residue number).
With keyword options:
stripPDB --input=in.pdb --output=out.pdb --remove=",,293"
With character options:
stripPDB -i in.pdb -o out.pdb -r ",,293"
The following will remove only the record(s) (and any associated CONECT records)
from 'in.pdb' that contains all of: chain identifier 'A', residue sequence number 35,
and atom serial number 293.
With keyword options:
stripPDB --input=in.pdb --output=out.pdb --remove="A,35,293"
With character options:
stripPDB -i in.pdb -o out.pdb -r "A,35,293"
The following will remove all the ATOM, HETATM, or TER records having:
chain identifier A, or
chain identifier B, or
both chain identifier C and a residue sequence number >= 1 and <= 49, or
both chain identifier C and a residue sequence number >= 63, or
chain identifier D.
With keyword options:
stripPDB --input=1BBT.pdb --output=temp1.pdb --from=“A;B;C,1-49;C,63-;D;”
With character options:
stripPDB -i 1BBT.pdb -o temp1.pdb -f “A;B;C,1-49;C,63-;D;”
LICENSE INFORMATION
stripPDB is a software program from Arthur Weininger (www.weiningerworks.com).
stripPDB is subject to a license; use the keyword option '--license' in order to view
the license terms. Your use of this software contitutes an agreement to the license
terms. Do not use this software if you do not agree to the license terms.
stripPDB Tutorial
The Picornavirus Monograph Superposition Shell Script gives examples of using stripPDB.