chainidPDB
Overview
chainidPDB Help Output
chainidPDB Tutorial
(WEB PAGE PDF)
chainidPDB Overview
chainidPDB is a PDB filter to change one or all chain identifiers.
chainidPDB Help Output (“chainidPDB -h” output)
NAME
chainidPDB (version 1.1.0) -- PDB filter to change chain identifiers
SYNOPSIS
chainidPDB [options]
CHARACTER OPTION_____KEYWORD OPTION________DESCRIPTION_____________________________DEFAULT__________
-i <filename> .... --input=<filename> ... input PDB file ........................ stdin
-a <model#> ...... --model=<model#> ...... MODEL # of input PDB file to process .. first model
-o <filename> .... --output=<filename> ... output PDB file ....................... stdout
-f <from_spec> ... --from=<from_spec> .... existing "from" target specification .. none
<from_spec> is either: <chain_ids> (simple list of chain ids with no ';' or ',')
or: [[c],[r],[a];]+ (triplet list of chain id, residue #, atom #)
-t <chain_ids> ... --to=<chain_ids> ...... replacement "to" chain identifiers .... required
-m <filename> .... --map=<filename> ...... generate file of atom changes ......... no map
-p ............... --nopositionchange .... do not change position of PDB records . positions changed
-x ............... --noresiduechange ..... do not change residue serial numbers .. residues changed
-h ............... --help ................ prints help (Enter 'chainidPDB -h' for help.)
<NO OPTIONS> ............................. shorter option synopsis (Enter 'chainidPDB'.)
--license ............. prints license terms for chainidPDB.
DESCRIPTION
chainidPDB is a PDB filter to change one or all chain identifiers.
chainidPDB reads an existing input PDB file and writes a new output PDB file.
Records matching a user-supplied target specification will have chain identifiers
replaced with user-supplied chain identifiers.
Input is expected to be in PDB format. Option '--input=' ('-i') specifies the input
filename. If option '--input=' ('-i') is not present, input will be read from stdin.
By default, the first MODEL (perhaps implicit if no MODEL records exist) from the
input file is processed. Option '--model=' ('-a') may be used to specify the MODEL
number of the input file to process.
Option '--output=' ('-o') specifies the output PDB filename. If option '--output=' ('-o')
is not present, output will be written to stdout.
New "REMARK 250" lines will be added to the output PDB specifying execution details.
By default, residues will be assigned new, unique residue serial numbers, new chains will
be output in a new chain order with changed atom records in new positions. By default,
a PDB file will be output that includes just the header records and specific ATOM, HETATM,
TER, and CONECT records for a single MODEL.
[Use of chainidPDB can easily produce invalid chains, which may be intended if
chainidPDB is being used to create temporary files with the chain identifiers
representing sets of atoms. Certain molecular visualization programs require unique
residue serial numbers within chains in order to display these residues and atoms.]
By default, chainidPDB fixes chains that have residues with duplicate residue
sequence numbers. Atoms will be written out sequentially by new alphanumeric chain order;
new atom serial numbers will be assigned as required. The new chain order will consist of
any specified chains in the following order:
'A' - 'Z'
'0' - '9'
'a' - 'z'
' ' (space)
< all other ASCII values from 1 to 127, inclusive >
Option '--noresiduechange' ('-x') may be used to specify that input residue numbers are to
remain unchanged; a warning will be generated when ambiguous residue numbers are output.
When the '--nopositionchange' ('-p') option is used, all PDB file records for the entire
MODEL being processed will be written to the output PDB file in the identical positions,
with respect to each other, of the input PDB file.
The option '--map=' ('-m') may be used to generate a text file listing atom changes.
No map file is generated by default.
Any errors and warnings will be written to stderr.
The option '--to=' ('-t') specifies the new, replacement chain identifiers as a simple
list of chain identifiers (e.g., "MNOP").
If the option '--from=' ('-f') is not present, then all ATOM, HETATM, and TER records
will be changed with each existing PDB chain identifier getting a successive user-
supplied replacement chain identifier.
If the option '--from=' ('-f') is present, then only ATOM, HETATM, and TER records
having content that matches the target specification of the '--from=' ('-f') option
value will be changed. The '--from=' ('-f') option value is either a simple list of
chain identifiers ("Simple List Format") or it is a list of triplets that allow chain
identifiers, residue sequence numbers, and atom serial numbers to be used to specify
matching record targets ("Triplet List Format").
Simple List Format
The Simple List Format of the '--from=' ('-f') option value is a character string of
chain identifiers. This character string may not contain semicolons (';') or commas (',')
unless the semicolons and commas are preceeded with a single backslash character ('\')
semicolons and commas not quoted by a backslash will cause the '--from=' ('-f') option
value to be interpreted as a Triplet List Format. Examples of the Simple List Format follow:
Terse option Keyword option Description
-------------- ---------------- -------------------------------------
-f ABC --from=ABC chain IDs 'A','B','C'
-f 'ABC XYZ' --from='ABC XYZ' chain IDs 'A','B','C',' ','X','Y','Z'
-f "ABC AAA" --from="ABC AAA" chain IDs 'A','B','C',' ','A','A','A'
Note that a single quote (') or a double quote (") may be used to delimit an option value.
The '--from=' ('-f') option values shown above are quoted in order to specify that a space (' ')
be used as a character identifier.
Note that use of the hyphen character ('-') in the Simple List Format will be interpreted as
the value of a chain identifier. See “Triplet List Format” below for range specifications.
Triplet List Format
The Triplet List Format of the '--from=' ('-f') option value allows records to be specified
for chain identifier replacement by presence of chain identifier, residue sequence number,
and atom serial number. This triplet list is defined as a semicolon-delimited list of
triplets, where each triplet consists of three comma-delimited fields:
chain identifier, residue sequence number, atom serial number
Any of the '--from=' ('-f') triplet fields may optionally have a range specification
in one of the following formats:
Format Examples Description
----------------------------- -------- --------------------------------------------
<lower_limit> - <upper_limit> A-Z Matches any value in between and including
107-200 lower limit and upper limit.
- <upper_limit> -Z Matches any value lower than and including
-200 upper limit.
<lower_limit> - A- Matches any value higher than and including
107- lower limit.
Any of the '--from=' ('-f') triplet fields may have a null value (i.e., an empty field).
Null-valued triplet fields will match any record value of that field type. A PDB record
has to match all non-null fields in a triplet in order for chain identifer replacement.
Comma delimiters need only be supplied as needed to specify a field index. Semicolon
delimiters are required with the exception of the last semicolon, which is optional.
Examples of the triplet list format follow:
Terse option Keyword option Description
-------------------- ---------------------- --------------------------------------------
-f 'A;B;C;' --from='A;B;C;' chain IDs (any residue # and any atom #)
-f 'A;B;C' --from='A;B;C' chain IDs (any residue # and any atom #)
-f 'A;B;C; ;X;Y;Z' --from='A;B;C; ;X;Y;Z' chain IDs (any residue # and any atom #)
-f "A;B;C; ;A;A;A" --from="A;B;C; ;A;A;A" chain IDs (any residue # and any atom #)
-f "A,,;" --from="A,,;" chain ID 'A' (any residue # and any atom #)
-f "A,35,;" --from="A,35,;" residue 35 of chain ID 'A' (with any atom #)
-f "A,35,293;" --from="A,35,293;" atom 293 of residue 35 of chain ID 'A'
-f ",35,;" --from=",35,;" residue 35 (any chain ID and any atom #)
-f ",,293;" --from=",,293;" atom 293 (any chain ID and any residue #)
-f "A,,;,35,;,,293;" --from="A,,;,35,;,,293;" three triplets with complete delimiters
-f "A;,35;,,293" --from="A;,35;,,293" three triplets with necessary delimiters
-r ",-78;,110-;" --from=",-78;,110-;" all residue #s except 79 through 109
Notes for both Simple List Format and Triplet List Format
The order of'--from=' ('-f') and '--to=' ('-t') target specifications is important in that:
(i) PDB records matching '--from=' ('-f') specifications get replacement chain
identifiers from identically indexed '--to=' ('-t') specifications, and
(ii) The first matching target specification will be used to change a record when
there are multiple triplets that specify identical records to be changed;
PDB records are only modified once.
If there are more replacement chain identifiers required than there are specified
'--to=' ('-t') chain identifiers, then the '--to=' ('-t') chain identifiers will be
reused; a warning will be given (to stderr) when this happens.
Chain identifiers, specified in '--from=' ('-f') or '--to=' ('-t') option values, can be
any non-NULL character. If semicolons (';'), commas (','), backslashes ('\'), or option
value quotes (''' or '"') are used as chain identifier values in any --from=' ('-f') or
'--to=' ('-t') option values then these characters need to be preceded with a single
backslash character ('\').
As the unix shell interprets semicolons (';') as command line separators, command line option
values containing semicolons (';') are best handled by quoting the entire option value with
either single quotes (') or double quotes (").
EXAMPLES
The following will change only the ATOM, HETATM, or TER records having the
chain identifier 'A' to similar records having the chain identifier 'B'.
With keyword options:
chainidPDB --input=infile.pdb --output=outfile.pdb --from=A --to=B
With character options:
chainidPDB -i infile.pdb -o outfile.pdb -f A -t B
The following will change all ATOM, HETATM, or TER records to similar records
having the chain identifier 'Z'. (No '--from=' ('-f') option was used.)
With keyword options:
chainidPDB --input=infile.pdb --output=outfile.pdb --to=Z
With character options:
chainidPDB -i infile.pdb -o outfile.pdb -t Z
The following will change only the ATOM, HETATM, or TER records having the
chain identifiers 'A', 'B', 'C', and 'D' to similar records having the
chain identifier '1', '2', '3', and '4', respectively.
With keyword options:
chainidPDB --input=infile.pdb --output=temp1.pdb --from="A;B;C;D" --to="1234
With character options:
chainidPDB -i infile.pdb -o temp1.pdb -f "A;B;C;D" -t 1234
The following will change only the ATOM, HETATM, or TER records having the
chain identifiers 'A', 'B', 'C', 'D', and ' ' (space) to similar records
having the chain identifier '1', '2', '3', '4', and 'Z', respectively.
With keyword options:
chainidPDB --input=infile.pdb --output=temp1.pdb --from="A;B;C;D; ;" --to="1234Z"
With character options:
chainidPDB -i infile.pdb -o temp1.pdb -f "A;B;C;D; ;" -t "1234Z"
The following will change only the ATOM, HETATM, or TER records having the
chain identifier 'A' and the residue number 36 to similar records having the
chain identifier 'Y'.
With keyword options:
chainidPDB --input=infile.pdb --output=temp1.pdb --from="A,36" --to="Y"
With character options:
chainidPDB -i infile.pdb -o temp1.pdb -f "A,36" -t "Y"
The following will change only the ATOM, HETATM, or TER records having the
residue sequence number 35 (and any chain identifier) to similar records having
the chain identifier 'Q'.
With keyword options:
chainidPDB --input=infile.pdb --output=temp1.pdb --from=",35" --to="Q"
With character options:
chainidPDB -i infile.pdb -o temp1.pdb -f ",35" -t "Q"
The following will change only the ATOM, HETATM, or TER records having the
atom serial number 293 (and any chain identifier or residue number) to similar
records having the chain identifier 'Z'.
With keyword options:
chainidPDB --input=infile.pdb --output=temp1.pdb --from=",,293" --to="Z"
With character options:
chainidPDB -i infile.pdb -o temp1.pdb -f ",,293" -t "Z"
The following example shows a triplet list having multiple triplet specifications
that may result in targeting identical records. The ATOM, HETATM, or TER records
having the chain identifier 'A' are specified to be changed to similar records
having the chain identifer 'X'. The ATOM, HETATM, or TER records having the
residue number 35 are specified to be changed to similar records having the chain
identifer 'Y'. The ATOM, HETATM, or TER records having the atom serial number 293
are specified to be changed to similar records having the chain identifer 'Z'.
With keyword options:
chainidPDB --input=infile.pdb --output=temp1.pdb --from="A;,35;,,293" --to="XYZ"
With character options:
chainidPDB -i infile.pdb -o temp1.pdb -f "A;,35;,,293" -t "XYZ"
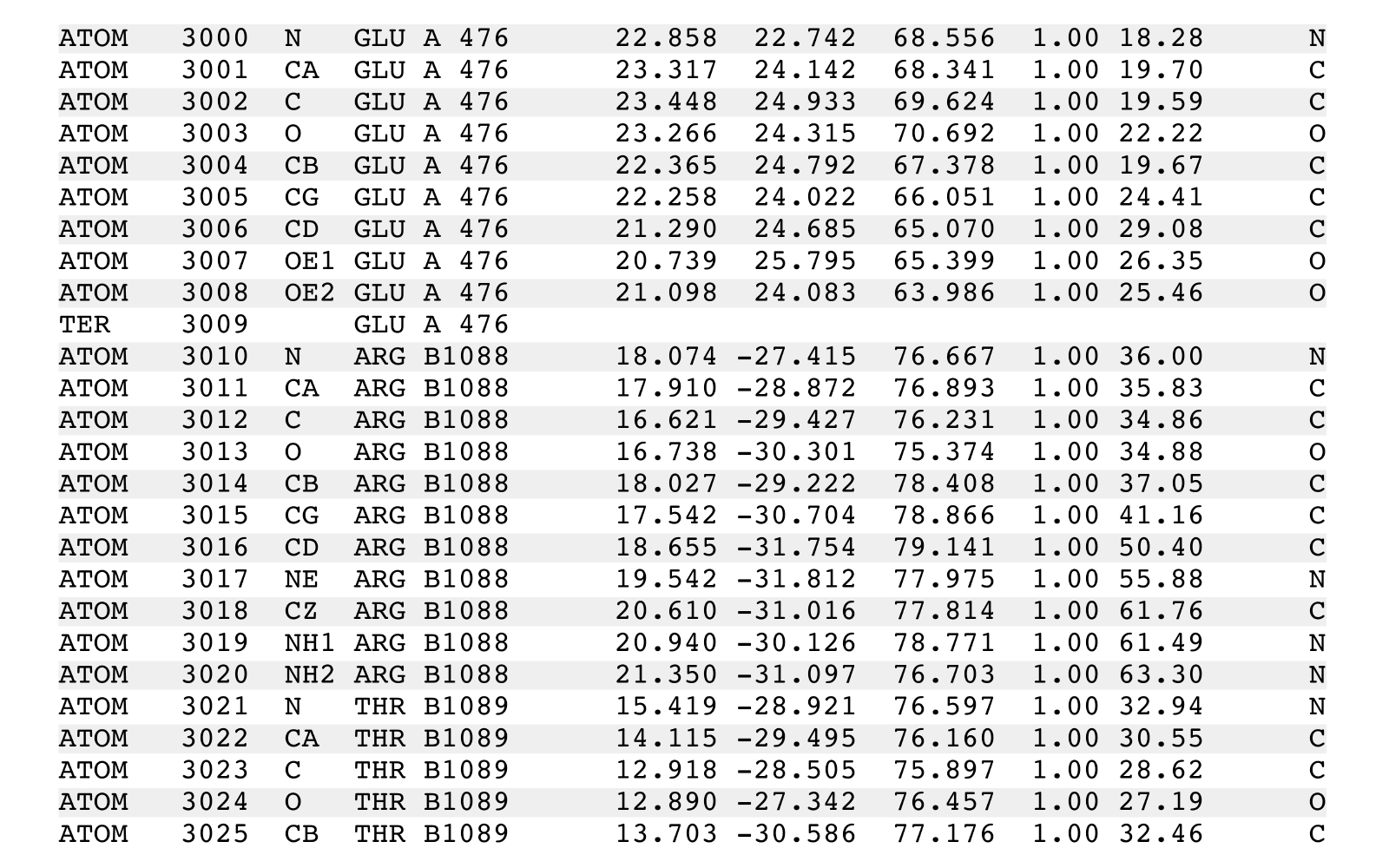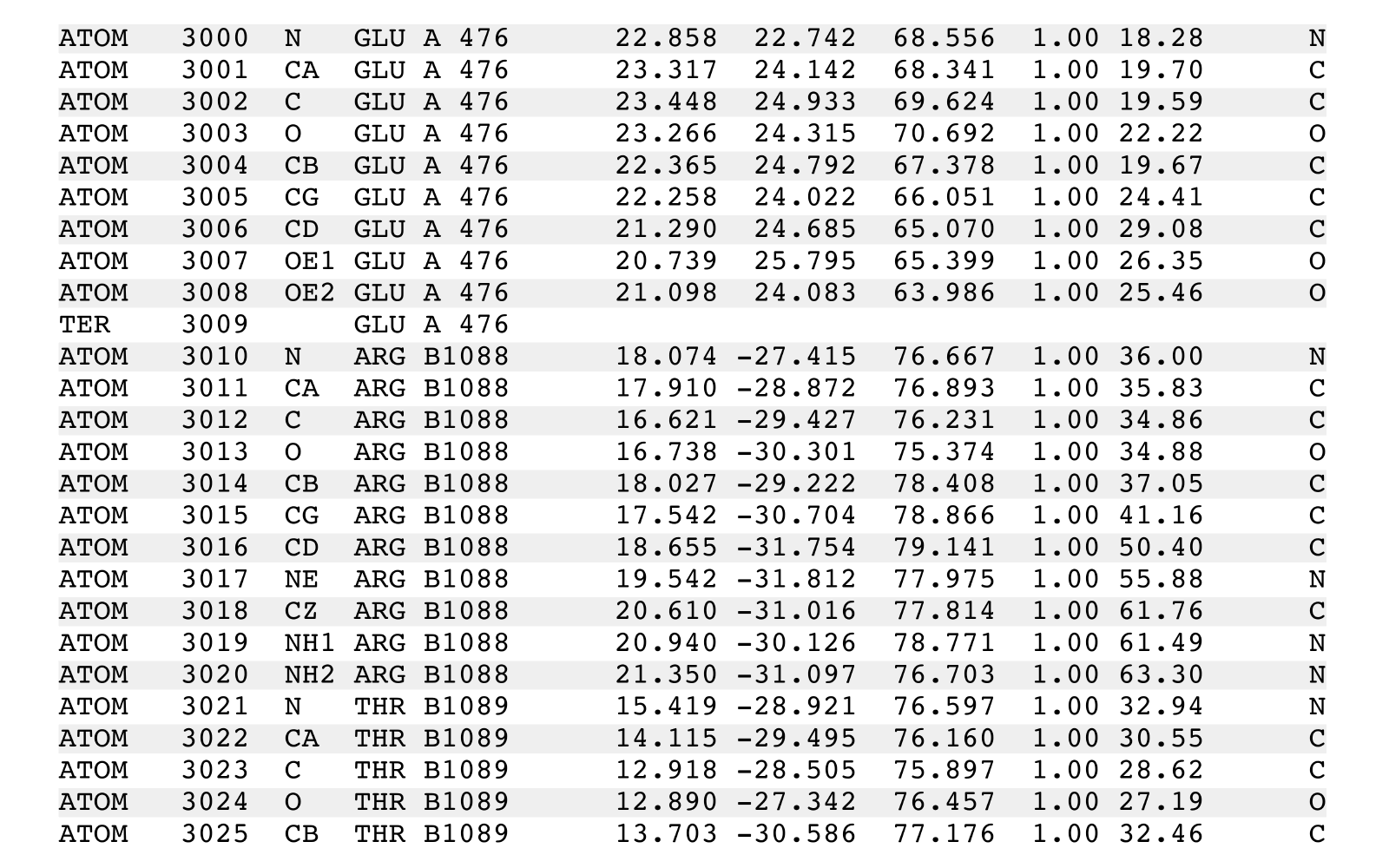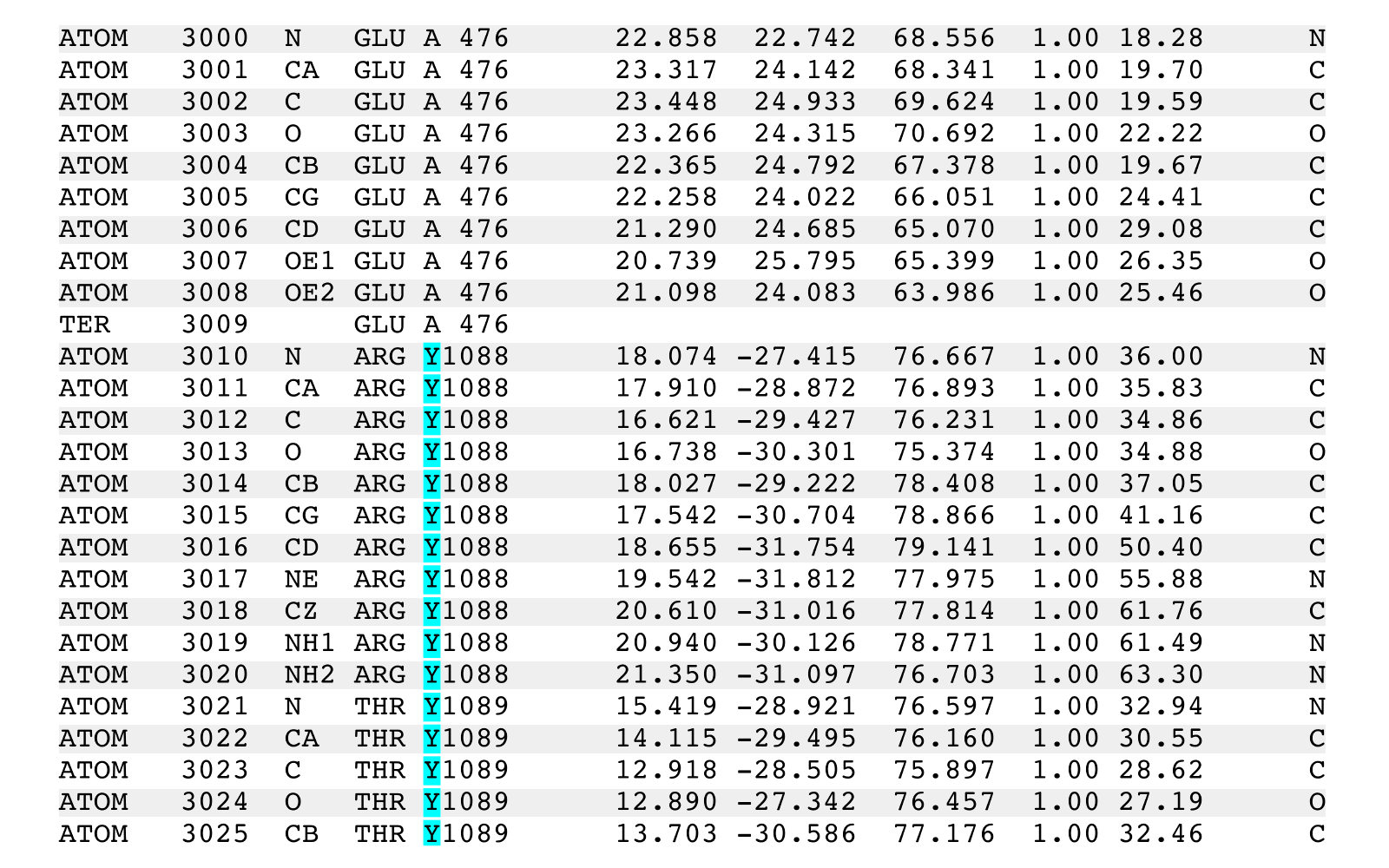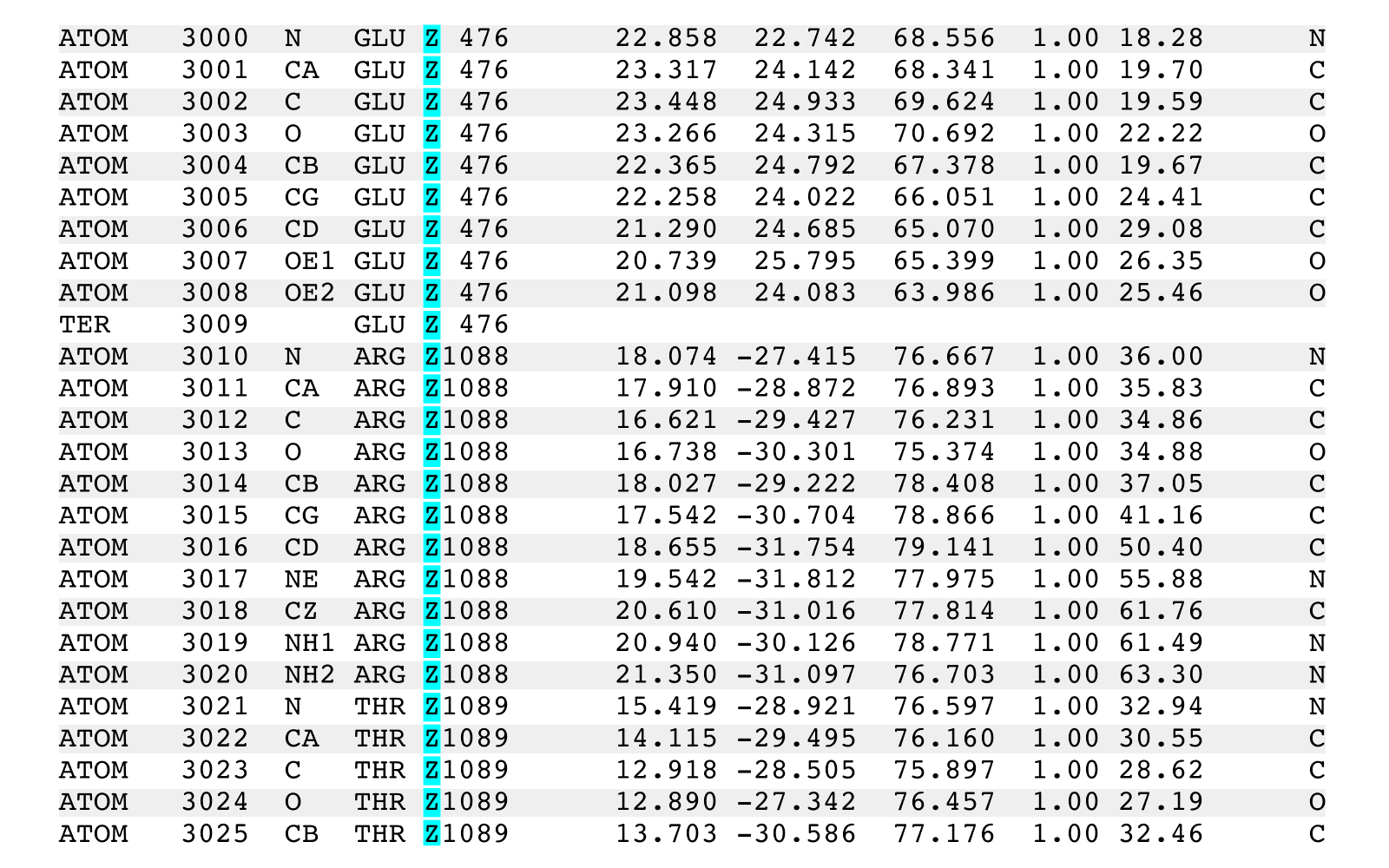
If the following was part of the input "infile.pdb" of the above chainidPDB cammand:
ATOM 287 CE2 TYR A 34 53.611 15.359 1.623 1.00 35.63 C
ATOM 288 CZ TYR A 34 54.658 15.283 2.521 1.00 36.11 C
ATOM 289 OH TYR A 34 54.406 15.463 3.861 1.00 35.18 O
ATOM 290 N ILE A 35 54.408 13.226 -4.218 1.00 38.28 N
ATOM 291 CA ILE A 35 54.395 13.169 -5.673 1.00 40.04 C
ATOM 292 C ILE A 35 53.691 14.397 -6.223 1.00 39.27 C
ATOM 293 O ILE A 35 52.849 15.000 -5.546 1.00 39.06 O
ATOM 294 CB ILE A 35 53.657 11.910 -6.210 1.00 41.31 C
ATOM 295 CG1 ILE A 35 52.229 11.858 -5.666 1.00 42.67 C
ATOM 296 CG2 ILE A 35 54.420 10.649 -5.838 1.00 42.60 C
ATOM 297 CD1 ILE A 35 51.227 12.665 -6.474 1.00 44.23 C
ATOM 298 N TRP A 36 54.062 14.775 -7.440 1.00 38.06 N
ATOM 299 CA TRP A 36 53.444 15.900 -8.121 1.00 37.24 C
ATOM 300 C TRP A 36 52.385 15.275 -9.024 1.00 37.32 C
Then the output would show that the PDB records having the chain identifier 'A' were
changed to similar records having the chain identifier 'X' unless the target records
had the residue number 35 in which case the output records were given the chain
identifier 'Y' unless the target record had the atom serial number 293 in which case
the output record was given the chain identifier 'Z':
ATOM 287 CE2 TYR X 34 53.611 15.359 1.623 1.00 35.63 C
ATOM 288 CZ TYR X 34 54.658 15.283 2.521 1.00 36.11 C
ATOM 289 OH TYR X 34 54.406 15.463 3.861 1.00 35.18 O
ATOM 290 N ILE Y 35 54.408 13.226 -4.218 1.00 38.28 N
ATOM 291 CA ILE Y 35 54.395 13.169 -5.673 1.00 40.04 C
ATOM 292 C ILE Y 35 53.691 14.397 -6.223 1.00 39.27 C
ATOM 293 O ILE Z 35 52.849 15.000 -5.546 1.00 39.06 O
ATOM 294 CB ILE Y 35 53.657 11.910 -6.210 1.00 41.31 C
ATOM 295 CG1 ILE Y 35 52.229 11.858 -5.666 1.00 42.67 C
ATOM 296 CG2 ILE Y 35 54.420 10.649 -5.838 1.00 42.60 C
ATOM 297 CD1 ILE Y 35 51.227 12.665 -6.474 1.00 44.23 C
ATOM 298 N TRP X 36 54.062 14.775 -7.440 1.00 38.06 N
ATOM 299 CA TRP X 36 53.444 15.900 -8.121 1.00 37.24 C
ATOM 300 C TRP X 36 52.385 15.275 -9.024 1.00 37.32 C
The above example was used to show multiple triplet specifications targeting identical
records. The triplet list used above ("A;,35;,,293") has a middle triplet (",35;")
that would change records having a residue 35 in ALL chains. The triplet ("A,35;")
would be used to only change records for residue 35 in chain A.
The following will change only the ATOM, HETATM, or TER records having:
chain identifier A, or
chain identifier B, or
both chain identifier C and a residue sequence number >= 1 and <= 49, or
both chain identifier C and a residue sequence number >= 63, or
chain identifier D.
The changed records will be given the chain identifer 'E'.
With keyword options:
chainidPDB --input=1BBT.pdb --output=temp1.pdb --from=“A;B;C,1-49;C,63-;D;” --to=“E”
With character options:
chainidPDB -i 1BBT.pdb -o temp1.pdb -f “A;B;C,1-49;C,63-;D;” -t “E”
LICENSE INFORMATION
chainidPDB is a software program from Arthur Weininger (weiningerworks.com).
chainidPDB is subject to a license; use the keyword option '--license' in order to view
the license terms. Your use of this software contitutes an agreement to the license terms.
Do not use this software if you do not agree to the license terms.
chainidPDB Tutorial
The Picornavirus Monograph Superposition Shell Script gives examples of using chainidPDB.